Prof. Li-Zhi Gao`s group (Plant Germplasm and Genomics Center, Germplasm Bank of Wild Species in Southwestern China, Kunming Institute of Botany, Chinese Academy of Sciences) and collaborators have made breakthrough on “The tea tree genome”, which provides insights into tea flavor and independent evolution of caffeine biosynthesis. (Xia et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Molecular Plant, 2017 DOI: 10.1016/j.molp.2017.04.002)

Kunming Institute of Botany researchers gathering tea tree leaves. Credit: Chao Shi
The following were extracted from the above paper:
Tea is the world’s oldest and most popular caffeine-containing beverage with immense economic, medicinal, and cultural importance. Here, we present the first high-quality nucleotide sequence of the repeat-rich (80.9%), 3.02-Gb genome of the cultivated tea tree Camellia sinensis. We show that an extraordinarily large genome size of tea tree is resulted from the slow, steady, and long-term amplification of a few LTR retrotransposon families. In addition to a recent whole-genome duplication event, lineage-specific expansions of genes associated with flavonoid metabolic biosynthesis were discovered, which enhance catechin production, terpene enzyme activation, and stress tolerance, important features for tea flavor and adaptation. We demonstrate an independent and rapid evolution of the tea caffeine synthesis pathway relative to cacao and coffee. A comparative study among 25 Camelliaspecies revealed that higher expression levels of most flavonoid- and caffeine- but not theanine-related genes contribute to the increased production of catechins and caffeine and thus enhance tea-processing suitability and tea quality. These novel findings pave the way for further metabolomic and functional genomic refinement of characteristic biosynthesis pathways and will help develop a more diversified set of tea flavors that would eventually satisfy and attract more tea drinkers worldwide.
Figure 1 Phylogenetic Analysis of the LTR Retrotransposon Sequences in the Tea Tree Genome.茶树基因组中LTR逆转录转座子序列的系统发育分析
The neighbor-joining and unrooted phylogenetic trees were constructed on the basis of 678 Ty1/copia(A) and 1795 Ty3/gypsy(B)aligned sequences corresponding to the RT domains without premature termination codon. LTR family names and proportion of each are indicated.
Figure 2 Evolution of the Tea Tree Genome and Gene Families.茶树基因组和基因家族的进化
(A) Venn diagram shows the shared and unique gene families among the tea tree and seven other plant species. Each number in parentheses represents the number of genes within corresponding families (without parentheses).
(B) Expansion and contraction of gene families among the 10 plant species. Phylogenetic tree was constructed based on 597 high-quality 1:1 single-copy orthologous genes using sacred lotus (Nelumbo nucifera) as outgroup. Pie diagram on each branch of the tree represents the proportion of genes undergoing gain (red) or loss (green) events. Number at root (13 476) denotes the total number of gene families predicted in the most recent common ancestor (MRCA) (see Supplemental Information). The numerical value beside each node shows the estimated divergent time of each node (myr).
(C) Comparisons of disease-resistant genes among the five plant species.
(D) Whole-genome duplication events detected in the tea tree.
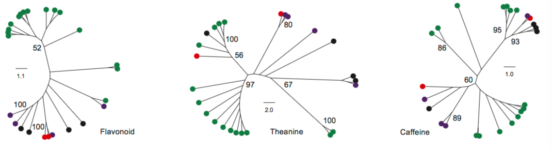
Figure 3 Evolutionary Differences of Three Important Metabolic Pathways Associated with Tea-Processing Suitability and Quality among the 25 Camellia Species. 与茶叶加工适应性和质量相关的三个重要代谢途径与25个茶树种的进化差异。
(A) Left panel represents the phylogenetic relationship of the 25 Camellia species constructed using whole-transcriptome sequencing data. Right panel shows the percent content of seven characteristic metabolites detected in the leaves of each Camellia species using HPLC (see Supplemental Information for abbreviation details).
(B) Expression profiles in FPKM (fragments per kilobase per million reads mapped) of key functional genes (rows) for each species (columns) related to three metabolic pathways in the tea tree. Data are plotted as log10 values. Right box plot indicates the expression correlations within section Thea (Thea; green), non-Thea sections (Non-Thea; orange), or between Thea and Non-Thea (gray).
(C) Sequence variations of genes involved in flavonoid, theanine, and caffeine metabolic pathways. Orthologous genes in each Camellia species were identified and multi-aligned to construct their phylogenetic topology.
Figure 4 Evolution of Caffeine Biosynthesis.咖啡因生物合成的进化
(A) The most essential and last three methylation steps for caffeine biosynthesis in plants. These methylation steps are catalyzed by a series of N-methyltransferases (NMTs), including xanthosine methyltransferase (7-NMT), theobromine synthase (7-methylxanthine methyltransferase; MXMT), and caffeine synthase (3, 7-dimethylxanthine methyltransferase; TCS). SAMS represents S-adenosylmethionine synthetase, while SAH indicates S-adenosylhomocysteine.
(B) Distribution of genes encoding NMTs among tea tree, coffee, and cacao genomes.
(C) Neighbor-joining (NJ) phylogenetic tree of NMT genes from tea tree (green solid dots), coffee (brown solid dots), and cacao (orange solid dots) NMTs. The 10 NMT genes cloned in seven wild relatives of tea tree, including C.irrawadiensis (green solid squares), C.ptilophylla (green solid lower triangles), C.granthamiana (green circles), C.lutchuensis (green squares), C.chrysantha (green diamonds), Camelliakissi (green lower triangles) and C.japonica(green upper triangles), are also shown and listed in Supplemental Table 36. The phylogeny shows high bootstrap support for independent evolution of the 13 caffeine biosynthesis genes.
(D) Expression profiles of 13 tea tree NMT genes (rows) based on RNA-seq data from eight different tissues (columns) (see Supplemental Information for names of abbreviated NMT genes).
Prof. Li-zhi Gao collecting tea tree leaves for sequencing. Credit: Yong-sheng Yi
The high content of catechins and caffeine detected in the tea tree and other species belonging to section Thea provide the fundamental basis of tea flavor. This provides possible answers to the basic biological question of tea-processing suitability and addresses, among hundreds of Camellia species, why favorable flavor generated by section Thea plants have long been appreciated, from which the cultivated tea tree C. sinensis was domesticated in human history, instead of those from non-Theasections. Our findings imply that wild relatives of cultivated tea tree present a huge reservoir for novel gene discovery toward the improvement of tea quality-related traits, whereas further studies are needed to dissect how differential expression of flavonoid and caffeine-related genes are related to regulating the accumulation of secondary metabolic compounds (Yan et al., 2015). The tea-processing industries in tea-drinking countries have developed numerous tea products with diverse tea flavor. Besides advances in tea-processing technologies, these depend on the development of a number of tea tree varieties containing diverse combinations of such characteristic secondary metabolites. We present an integrative data framework based on large-scale phytochemical, transcriptomic, and functional data, which will enable further metabolomic and functional genomic refinement of characteristic biosynthetic pathways including secondary metabolites, to form a more diversified set of tea flavors that will eventually satisfy and attract more tea drinkers worldwide. Considering the rapid extinction and severely endangered status of natural wild tea tree populations due to leaf over-harvesting creating tea products at high market prices (Liu et al., 2012), the genome assembly of the tea tree and transcriptomic variation data presented here will offer valuable information to aid the global conservation of these precious wild tea tree species.
Prof. Gao is now on the Advisory Board of World Tea Organization. For more information WTeaO.org, please feel free to contact [email protected] 001-2253041387 or http://wteao.org/contact/.
关于世界茶叶组织更多资讯与合作,欢迎联系WTeaO.org [email protected],微信:drtea1 或者 http://wteao.org/contact/




Comments by wteao
Prof. Shen Dongmei`s New Book on Tea
Rajiv Rochan---“ambassador of tea” of India in China, India Director ...